
Спінальна м'язова атрофія – важка патологія, яка часто робить прості дії, такі як ходіння, сидіння, недоступними для дитини. Малюк може виявитися позбавлений навіть такої природної можливості, як самостійне дихання. Прогнози при даній патології робити дуже складно, адже ні спеціального лікування, ні профілактики не існує, а все залежить від форми захворювання і факторів, яким медицина пояснення знайти не може.

Що це?
Говорячи про спінальної м'язової атрофії, мають на увазі не одне конкретне захворювання, а цілу групу хвороб під спільною абревіатурою СМА. Всі вони спадкові і пов'язані з дегенерацією нервових клітин спинного мозку, що відповідають за рухові функції.
Серед генетичних патологій у дітей спинальні м'язові атрофії займають лідируючі місця по частоті поширення. І приблизно один з 6 тисяч дітей народжується на світ з таким страшним діагнозом. У 50% випадків діти не доживають до дворічного віку і гинуть. Життя інших – це інвалідність.


Проблема, на думку генетиків, куди ширше, ніж може здатися з наведеної статистики.
Захворювання розвивається через мутації певних генів, і один з них – SMN1, який вважається головним «винуватцем» патології, у видозміненому вигляді рецессивно присутня у кожного п'ятдесятого жителя планети. А це означає, що у здорових батьків, які навіть не здогадуються про те, що є рецесивними носіями мутованого гена, цілком може з'явитися на світ малюк зі спінальною м'язовою атрофією.
Група хвороб вперше була описана в XIX столітті Гвідо Вердингом, ім'ям якого потім була названа одна з різновидів дитячих СМА.

Класифікація
Найпоширенішою формою СМА у дітей є проксимальна. Вона представлена кількома видами захворювання, не всі з яких стають очевидними відразу після народження дитини.
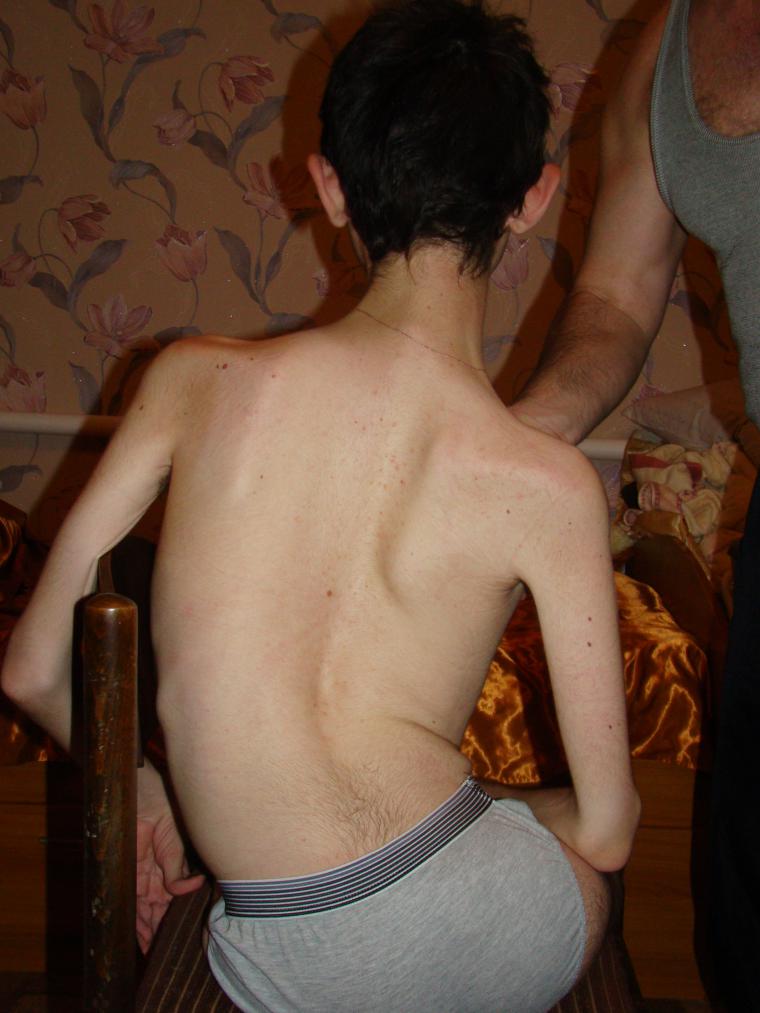
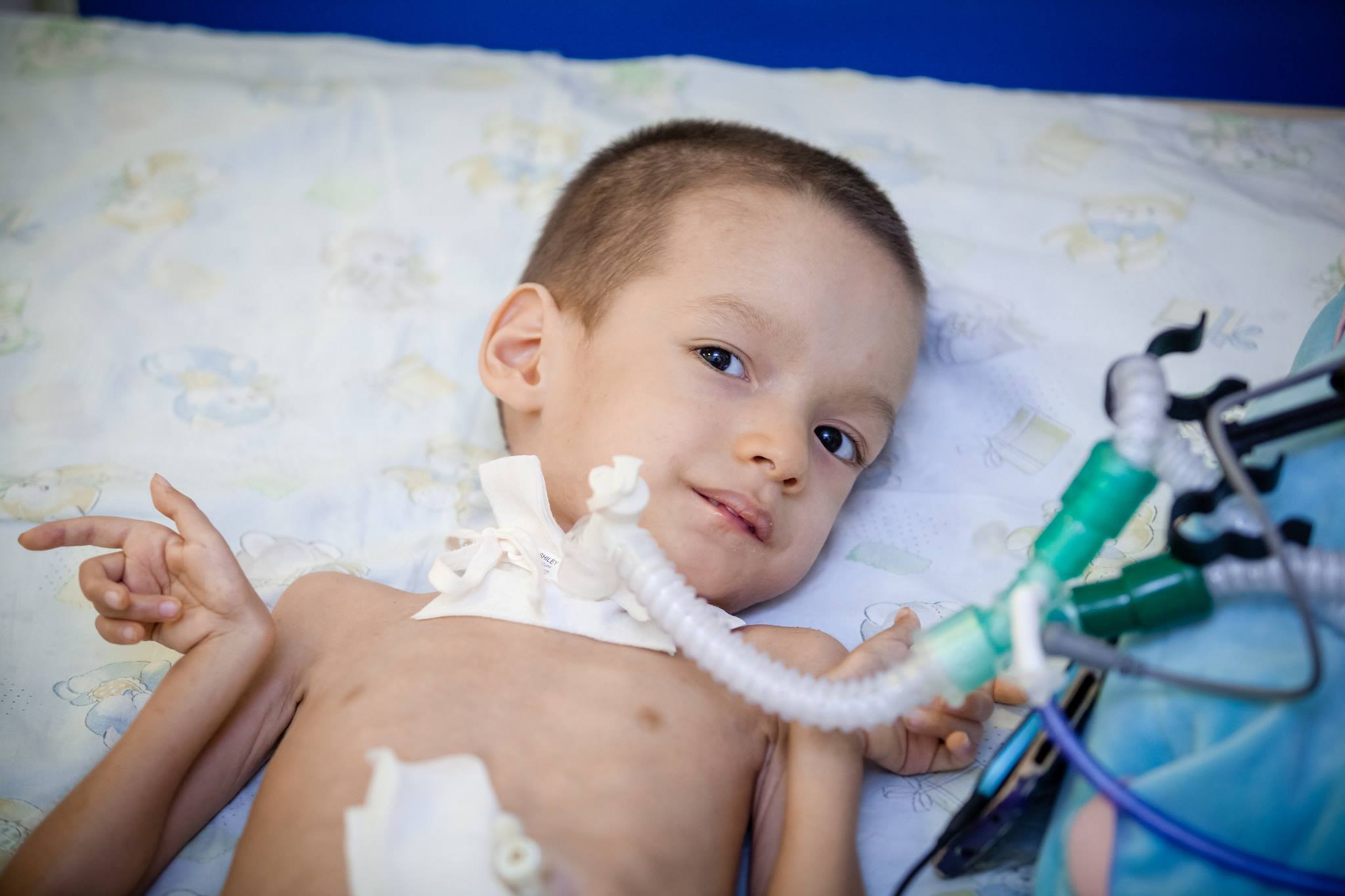
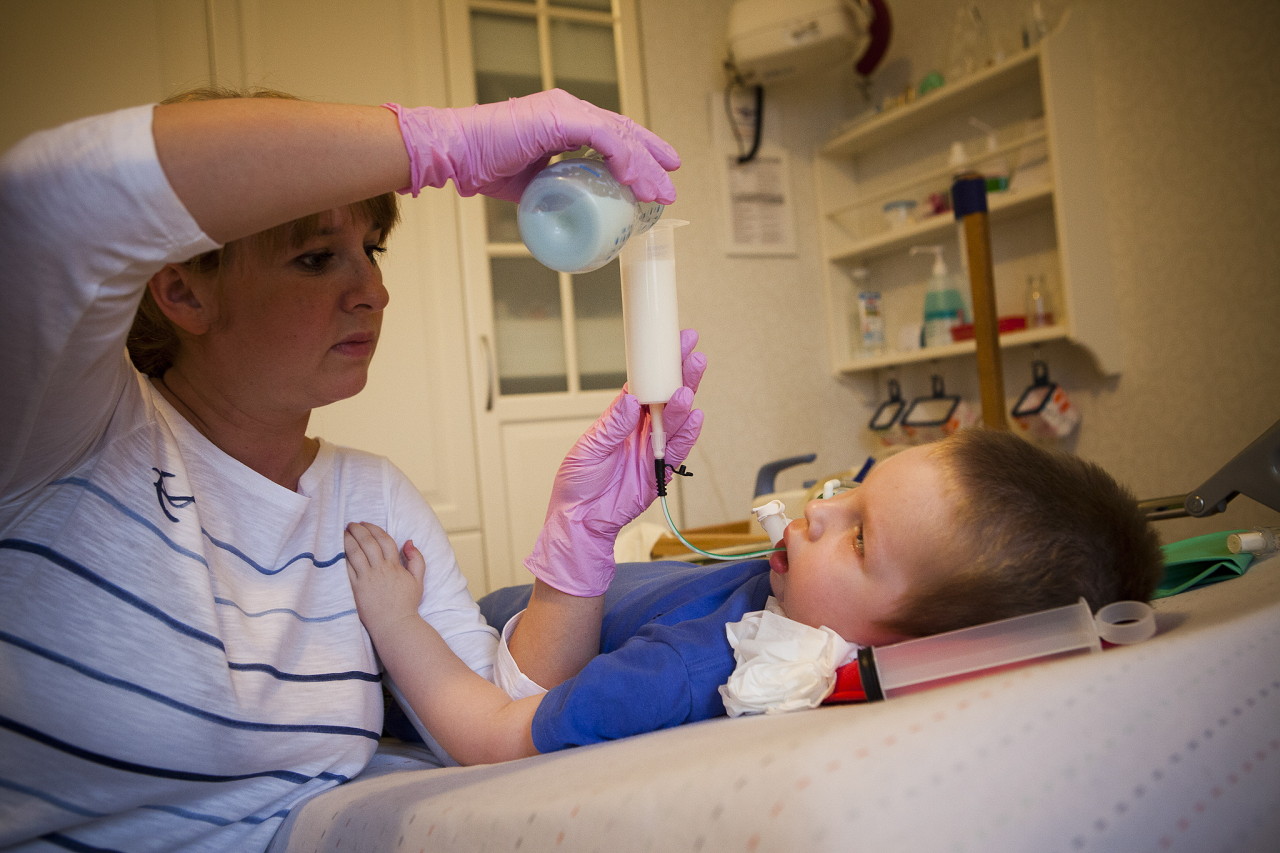
- Хвороба Вердинга-Гоффмана – СМА 1 типу, важка дитяча хвороба, яка проявляється в перші півроку життя дитини. Прогнози при ній найнесприятливіші, більшість пацієнтів гинуть. Дитина з СМА 1 типу не може ні стояти, ні сидіти, ні самостійно перевертатися. У багатьох новонароджених порушені смоктальні і ковтальні рефлекси. Часто відсутня можливість самостійного дихання або дихання утруднене.
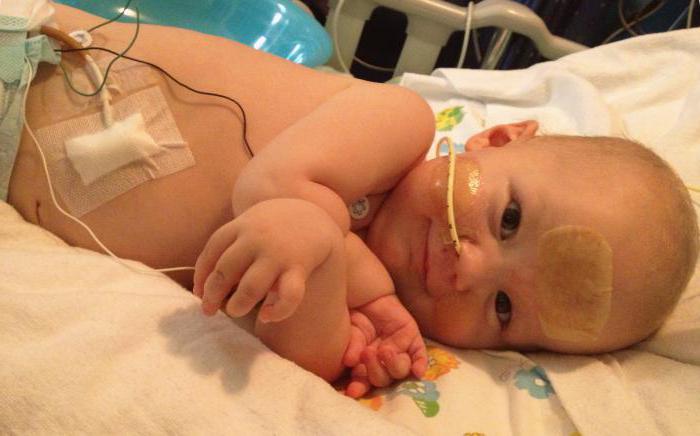
- Атрофія Дубовиця – СМА 2 типу, пізня дитяча. Виявляється зазвичай у віці від півроку до півтора років і пізніше. Ходити, стояти дитина не може, але здатний сидіти, харчування не порушено, він цілком справляється із завданням ковтання, смоктання. Скільки проживе малюк, залежить від того, в якому стані знаходяться дихальні м'язи.
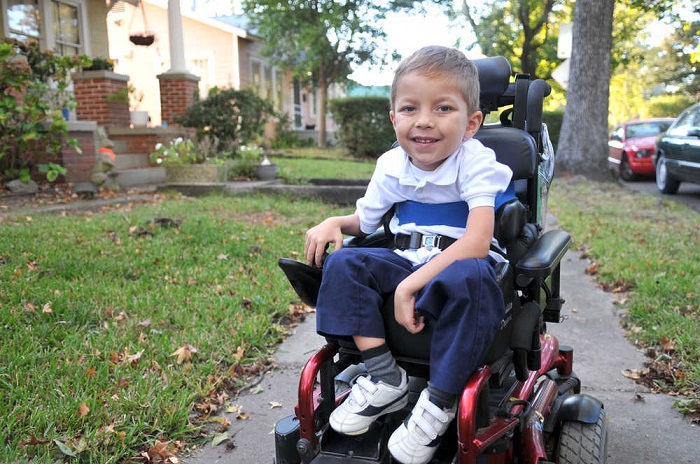
- Атрофія Кугельберга-Веландер – СМА 3 типу, інфантильна. Зазвичай виявляється у віці від півтора років, частіше у два роки. Прогностично більш сприятлива форма. Маленькі пацієнти можуть стояти, сидіти, переміщатися, але відчувають сильну слабкість, а тому в більшості випадків потребують в інвалідному кріслі, без якої нормальна життєдіяльність для них утруднена.
- Атрофія Кеннеді – СМА 4 типу, бульбоспинальная. Зазвичай вважається дорослою формою, але зрідка виявляється і у дітей після 15 років. На тривалість життя впливає рідко, ослаблення м'язів відбувається повільно, поступово, людина, який вів звичайне життя, і вважав себе здоровим, з часом стає інвалідом і втрачає здатність пересуватися самостійно.


Більш-менш відома чуток атрофія Дюшена та атрофія Вюльпиана – СМА «дорослі», перша виявляється зазвичай після 18 років, а друга – після 20 років.
У дітей реєструються не тільки ізольовані форми СМА, коли, крім дистрофії м'язів, нічого не турбує, але і поєднані форми, коли спінальна атрофія – не єдиний діагноз у дитини є інші генетичні або вроджені проблеми, наприклад, вади серця та судин, олігофренія.

Причини
Як вже мовилося, мова йде про генетичне захворювання, а тому причини його виникнення – область пошуків генетиків. Дитина успадковує один з рецесивних генів на п'ятій хромосомі (це можуть бути гени SMN, NAIP, H4F5, BTF2p44).
Ймовірність передати нащадкам такий ген у носія висока – 25%. Якщо і мама, і тато – приховані носії мутованого гена, то ймовірність СМА у дитини – 50%. Вражений аномальний ген не дає нормально протікати процесів вироблення білка SMN і нервові клітини, що відповідають за рухові функції м'язів, у спинному мозку починають поступово гинути. Процес їх загибелі продовжується і після того, як малюк з'явиться на світ.
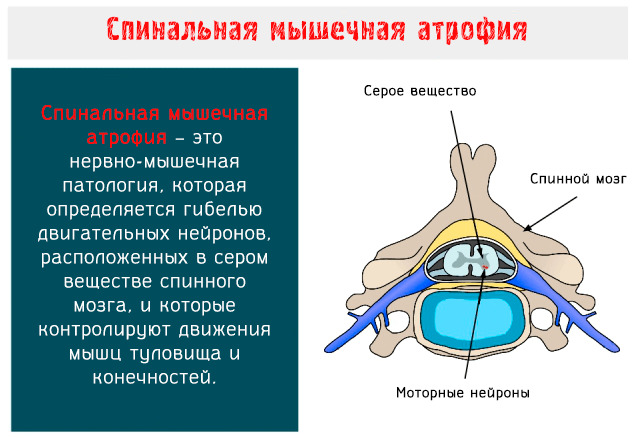
Прояви
Симптоми залежать від типу хвороби. Оскільки ми розглядає тільки дитячі чотири типу, то слід зазначити, що для всіх характерна м'язова слабкість, м'язова атрофія. В іншому кожен тип має власну клінічну картину та відмінні особливості.
- СМА 1 типу (атрофія Вердинга-Гоффмана) доступна для виявлення ще під час вагітності. Запідозрити захворювання у плода лікар може при дуже млявих шевелениях. Але підтвердити діагноз на етапі виношування дитини складно, це зазвичай відбувається після пологів. Малюк з такою атрофією не може сам тримати голівку, перевертатися з боку на бік, не сідає. Він майже постійно лежить на спинці, його поза розслаблена, він не піднімає ніжки, не зводить їх разом, не складає разом долоньки рук. На самому ранньому етапі можуть бути величезні проблеми з тим, щоб нагодувати дитину, адже ковтати у нього виходить дуже погано або не виходить. Велика частина діток гине ще до дворічного віку. Деяким вдається дожити до семи-восьми років, але атрофія тільки посилюється. Зазвичай загибель відбувається із-за недостатності роботи серця, легенів, травних органів.
- СМА 2 типу (атрофія Дубовиця) при народженні зазвичай не виявляється, адже дитина здатна дихати, ковтати їжу, і тільки після півроку стає очевидним прогрес м'язової атрофії. Якщо перші симптоми припадають на вік, коли дитина вже навчився стояти в ліжечку, то яскравою ознакою може бути подкашивание ніжок, безпричинні падіння крихітки. Поступово йому стає важко ковтати. З часом дитина починає потребувати в інвалідному кріслі.
- СМА 3 типу (аміотрофія Кюгельберга-Веландера) може виявитися в будь-якому віці після 2 років до повноліття. Дитина, яка нормально ріс і розвивався, поступово починає скаржитися на слабкість, зазвичай в області плечей, передпліч. По мірі прогресування йому стає важко бігати, ходити по сходах, присідати. Все залежить від догляду – деякі зберігають здатність переміщатися самостійно протягом довгих років.
- СМА 4 типу (атрофія Кеннеді) зустрічається тільки у пацієнтів чоловічої статі, оскільки вважається зчепленої з статевою хромосомою Х. Перші ознаки – слабкість в області стегнових м'язів, поступово уражаються черепні нерви. Захворювання прогресує повільно.

Лікування
На жаль, на сьогоднішній день медична наука не може запропонувати методів і засобів для лікування СМА. Таких методів не існує. Для підтримки функцій організму і максимального подовження періоду, поки дитина може рухатися сам, рекомендуються такі медикаменти, як «Прозерин», «Оксазил». Вони знижують активність ферменту, здатного розщеплювати ацетилхолін, передає імпульс збудження по волокнам нервової системи.
Також рекомендується систематичний прийом засобів, що підсилюють енергообмін на клітинному рівні, вітамінів групи В, ноотропних засобів, а також препаратів калію і нікотинової кислоти.
Дитині з СМА показано дотримання дієти з високим вмістом білка, але останні дослідження показали, що роль дієти дещо перебільшена – немає доказів, що великий вміст протеїну в їжі хоч якимось чином впливає на швидкість прогресування недуги.
А ось з калоріями слід бути більш уважними – з-за зниженою активності м'язів дитина швидко може набрати зайві кілограми.
Допомогти продовжити період більш-менш повноцінного життя допоможуть лікувальний масаж, УВЧ, електрофорез, програми дихальної гімнастики для підтримки дихальних м'язів, плавання. Рекомендується носіння підтримуючих спинних і грудних ортопедичних пристосувань.

Детальніше про хвороби розповідає фахівець у відео, представленому нижче.